
Reproducibility in macromolecular crystallography (MX) is primarily challenged by the complexity of data analysis workflows. The progression from raw X-ray detector data to refined structures involves a series of different software and parameter choices that can substantially affect the final result. Specific decisions regarding resolution cutoffs, space groups, and other refinement settings are frequently revisited as data interpretation evolves, especially when responding to reviewer requests or testing new hypotheses.
As a result, reproducible research in MX depends not only on preserving final outputs but also on maintaining a transparent data analysis history. This requires clear records of the software versions used, the inputs provided, the chosen parameters, and the manner in which results changed when those settings were adjusted. Consistent execution environments are an important part of this record. They ensure that rerunning a workflow produces reproducible results even as software evolves or members in a research group change. This reliability becomes particularly important when working at low resolution, where statistical error is more difficult to interpret and alternative processing or refinement strategies are routinely explored.
DECTRIS CLOUD offers a practical way to address these challenges by replacing person-dependent workflows with deterministic execution. The exact execution environment for a specific analysis result, down to the operating system and all software dependencies, can be revisited years later with a click of a button without having to rebuild it. In this article, we walk through a set of public job templates covering three common stages of MX data analysis: integration of peak intensities, molecular replacement, model building and refinement. For each step, we highlight how reproducibility is supported in practice by making input parameters visible, enabling straightforward reruns with modified settings, and linking final results back to the complete computational workflow that produced them.
Integration of peak intensities
The first stage of macromolecular crystallography (MX) data analysis transforms raw diffraction images into integrated reflection intensities. This process involves several linked steps: identifying diffraction spots, indexing the crystal lattice, refining experimental geometry, and integrating intensities across the crystal rotation range. While these steps are often automated, they rely on parameters that directly influence data quality.
Several widely used software packages are available for diffraction image processing, most notably DIALS [1] and XDS [2, 3]. In practice, many researchers interact with these tools through higher-level wrappers that standardize workflows, such as xia2 [4] (which provides a unified interface to multiple backends) and Fast DP [5, 6, 7] (optimized for rapid processing during data collection).
DECTRIS CLOUD provides example public job templates for these workflows, ensuring a consistent and reproducible environment:
- PRETTY XDS: An XDS-based workflow where you provide your own input files for xds_par and xdsconv for maximum control.
- DIALS example job: Diffraction image processing using the DIALS framework
- xia2 example job: Automated integration using xia2, which can be both based on XDS and DIALS.
- Fast DP job: Rapid integration and assessment during data collection with XDS.
With these templates, integration workflows can be launched and relaunched with full visibility into all input parameters. A common use case is adjusting the high-resolution cutoff (d_min) after evaluating initial data quality. In the accompanying video, a xia2 job is rerun with a modified resolution limit while all other conditions remain identical.
Video: Example of running the xia2 example job on DECTRIS CLOUD, inspecting the result, and adjusting the parameters in a new job rerun.
The resulting changes in statistics, such as CC1/2 and ⟨I/σ(I)⟩, can be inspected, making it straightforward to assess the impact of a single parameter choice. Crucially, each run preserves its complete analysis context. This enables direct comparison between alternative strategies without overwriting earlier results. The integrated reflection files produced here form the foundation for subsequent steps, such as molecular replacement, where the consequences of these early choices become fully apparent.
Molecular replacement
Once raw diffraction data has been integrated and scaled, the next challenge in the MX pipeline is the phase problem. Because X-ray detectors record only the intensities of the reflections and not their phases, the phase information essential for reconstructing the electron density map must be recovered through other means.
Molecular Replacement (MR) is the most common method for overcoming this hurdle when a structural model of a related protein (a homologoue) is already known. The process involves taking a previously solved structure, known as the search model, and computationally rotating and translating it within the unit cell of the new crystal. The goal is to find the orientation and position that best accounts for the observed diffraction pattern. When successful, the phases from the search model are used as an initial estimate for the new structure. This provides a starting point for subsequent model building and refinement.
On DECTRIS CLOUD, we provide dedicated example job templates for this stage using MOLREP [8], which is a highly robust and long-standing program within the CCP4 suite.
- Molecular Replacement MOLREP: This template automates the standard execution of the MOLREP program. It allows users to provide their integrated reflection data in MTZ format along with a coordinates file of the search model in PDB format. It is designed for straightforward cases where a single model or domain needs to be placed into the density.
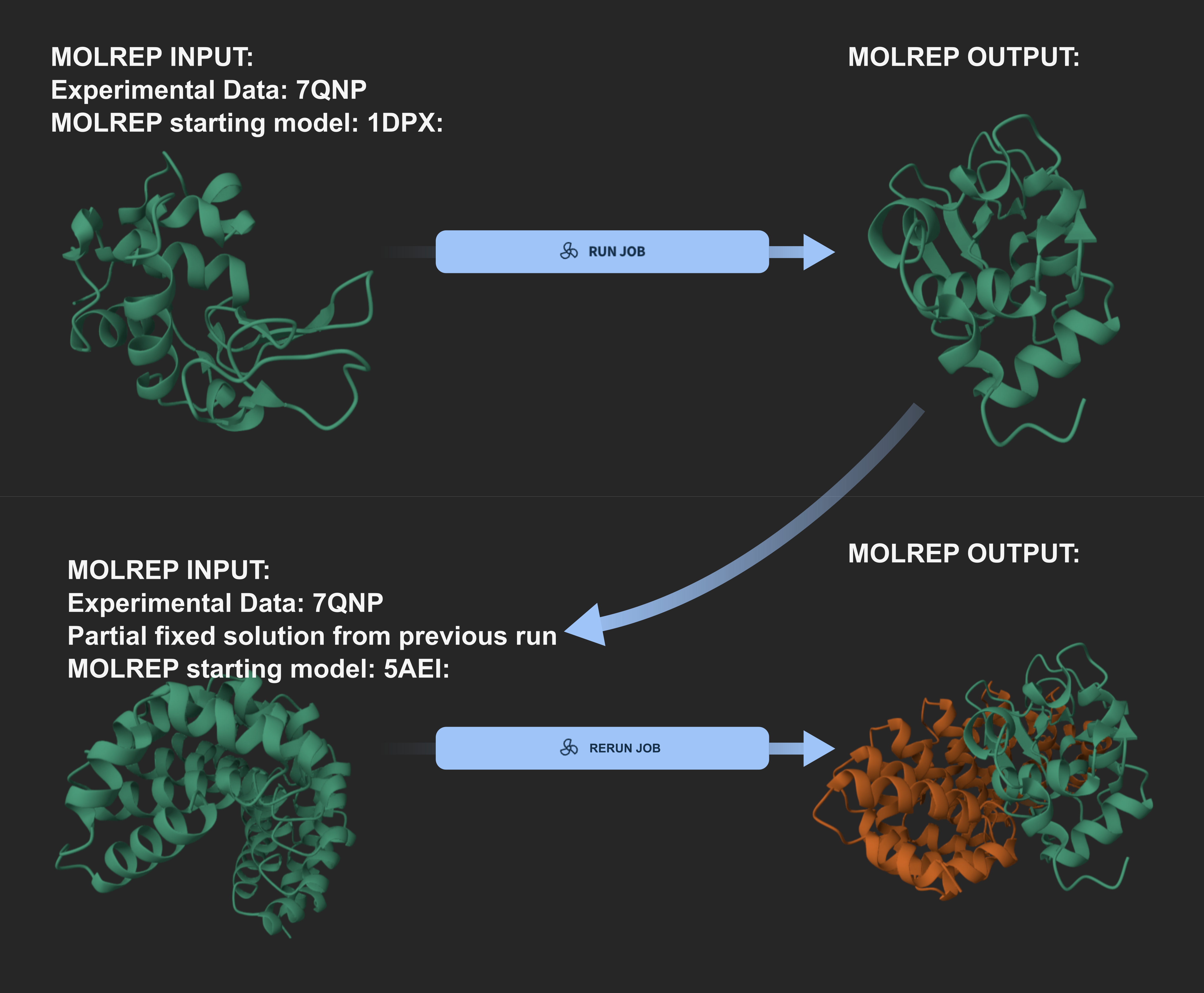
- Sequential MOLREP: This template allows for more advanced configuration of the starting parameters. Users can define the starting model via a PDB code or by providing a path to a custom prepared model. Crucially, this template supports an iterative workflow by allowing the user to provide a path to a partial solution. This partial model remains fixed in place while the program searches for the next component, which is a critical feature when solving structures with multiple subunits or distinct domains. By performing several sequential MR runs, users can build up complex assemblies piece by piece, ensuring each new subunit fits correctly around those already placed.
This transparent handling of molecular replacement is especially valuable when workflows are shared between collaborators or revisited at a later stage of the project. In complex cases where multiple sequential runs are required to place various subunits, the system maintains a clear lineage of the structural solution. Instead of overwriting previous attempts, each molecular replacement task is recorded as a unique event in the project timeline. By linking these results back to the integrated data and the specific parameters used in each step, researchers can explore alternative hypotheses without losing the previous analysis history.
Structural Refinement and Model Building
The transition from a molecular replacement solution to a finalized structure is an iterative process of automated refinement and manual model building. This stage bridges the gap between the initial search model and the experimental diffraction data.
Using REFMAC5 [9], the atomic coordinates and B-factors of the model are mathematically optimized to maximize their agreement with the experimental electron density, while simultaneously applying restraints to maintain chemically valid bond lengths, angles, and planes.
- REFMAC5 example job: This template executes the refinement of protein coordinates against integrated reflection data. It automates the optimization of the atomic model and calculates the difference electron density maps and final R-factors (R-work and R-free), which serve as the primary statistical indicators of model quality. The job generates the refined atomic models in both pdb and mmcif formats. The inclusion of cif files ensures the output is ready for modern deposition standards.
In a typical workflow, the initial molecular replacement solution can first be processed through the REFMAC5 job template to generate the necessary electron density maps. If necessary, a researcher can then proceed to manual model building, where discrepancies between the experimental data and the model are solved by hand. This includes specialized tasks such as fitting ligands into unexplained density, adding water molecules, or correcting side-chain rotamers to better match the map.
In DECTRIS CLOUD, these manual workflows are supported via dedicated virtual machines (sessions). These can be started within a compute environment with the CCP4 suite pre-installed, allowing researchers to perform manual building using Coot [10, 11] without needing the software or data files located on their own local computer. Following this manual model building, the REFMAC5 job can be re-run with the improved coordinates to perform a final optimization and update the statistical indicators of the model.
In order to ensure full transparency, the REFMAC5 job mentioned above copies the input PDB file to its output directory. This practice ensures that the starting point of the job run is clear and that the transition from manual intervention to automated optimization is fully documented.

Scripted Visualization
A common way of visualizing the final output of an MX workflow is via Coot [10, 11]. As mentioned previously, this can be done within the DECTRIS CLOUD virtual machines, providing a high-performance environment for interactive model building and validation.
However, visualization can also be automated to assist in rapid data assessment. It is possible to script Coot to generate standardized views or snapshots, as demonstrated in the following public job template:
- Coot script example: This template automatically visualizes PDB and MTZ files and generates zoomed-in views of the regions with the largest density differences.
This job template can be integrated into a more automated pipeline. By automatically generating images of the most significant features or discrepancies in a map, researchers can quickly evaluate the final result of a refinement run without manually opening every file. This step ensures that even the final visual assessment is part of a consistent, documented workflow.

Conclusion and Cloud Consumption Cost
Reproducibility in macromolecular crystallography is not achieved by a single software choice, but through the transparent management of the entire analysis pipeline. By using job templates on DECTRIS CLOUD, researchers move beyond isolated log files and fragile, fragmented local directories. Instead, they capture the complete computational state of their work. These workflows ensure that every parameter choice is recorded and that exact starting points from manual analysis are preserved. This provides a fully deterministic and traceable path from raw diffraction data to a refined PDB entry, empowering labs to rerun analyses on demand.
Because MX data processing is highly computationally efficient, these workflows can often be tested and carried out on a free account due to their low resource consumption. To provide an overview of expected performance, the table below shows example execution times for several public datasets using our standard job templates:
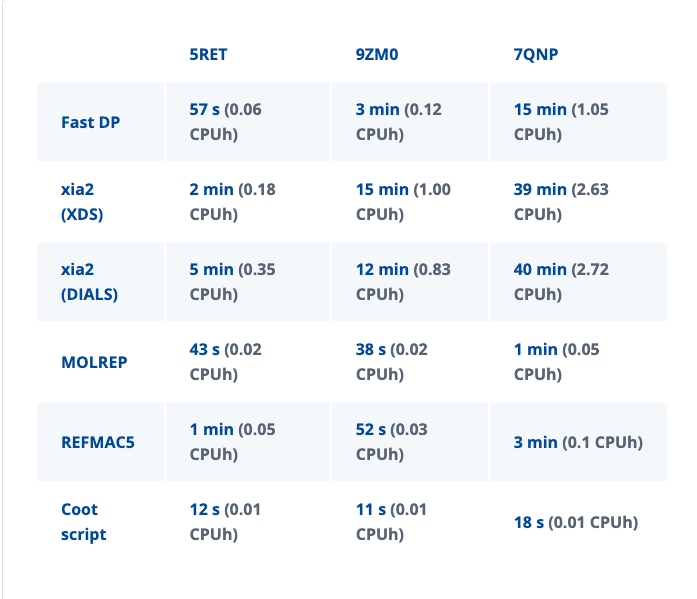
With the standard 20 CPUh included per month in a free license, it is possible to explore these workflows in several iterations. As an example, the xia2 (XDS) job took 2 min and 41 s to run for the 5RET dataset on a 4 CPU node. This is converted to CPUh via the calculation:
Job run time X #CPU = (2 min 41 s) X 4 CPU = (0.045 h) X 4 CPU = 0.18 CPUh
Meaning that the job run time can be converted into 0.18 CPUh for this node type. As such, a free account would be able to run this analysis workflow more than 100 times each month without exceeding its limits.
Interested in trying out MX workflows on DECTRIS CLOUD yourself? All workflows described above are freely accessible for any academic user with a free account.
References
- DIALS: implementation and evaluation of a new integration package. Winter G, Waterman DG, Parkhurst JM, Brewster AS, Gildea RJ, Gerstel M, Fuentes-Montero L, Vollmar M, Michels-Clark T, Young ID, Sauter NK, Evans G (2018). Acta Crystallogr D Struct Biol 74, 85-97
- XDS. Kabsch W (2010). Acta Crystallogr D Biol Crystallogr 66, 125-132.
- Integration, scaling, space-group assignment and post-refinement. Kabsch, W. (2010). Acta Crystallographica Section D, 66(2), 133–144.
- Decision making in xia2. Winter G, Lobley CMC, Prince SM (2013). Acta Crystallogr D Biol Crystallogr 69, 1260-1273.
- Automated data collection for macromolecular crystallography. Winter G, McAuley KE (2011). Methods 55, 81-93.
- The Computational Crystallography Toolbox: crystallographic algorithms in a reusable software framework. Grosse-Kunstleve RW, Sauter NK, Moriarty NW, Adams PD (2002). J Appl Crystallogr 35, 126-136.
- Overview of the CCP4 suite and current developments. Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, et al. (2011). Acta Crystallogr D Biol Crystallogr 67, 235-242
- MOLREP: an automated program for molecular replacement. Vagin A, Teplyakov A (1997). J Appl Crystallogr 30, 1022-1025.
- REFMAC5 for the refinement of macromolecular crystal structures. Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA (2011). Acta Crystallogr D Biol Crystallogr 67, 355-367.
- Coot: model-building tools for molecular graphics. Emsley P, Cowtan K (2004). Acta Crystallogr D Biol Crystallogr 60, 2126-2132.
- Features and development of Coot. Emsley P, Lohkamp B, Scott WG, Cowtan K (2010). Acta Crystallogr D Biol Crystallogr 66, 486-501.
- 5RET Data: Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. Douangamath A, Fearon D, Gehrtz P, Krojer T, Lukacik P, Owen CD, Resnick E, Strain-Damerell C, Aimon A, Abranyi-Balogh P, Brandao-Neto J, Carbery A, Davison G, Dias A, Downes TD, Dunnett L, Fairhead M, Firth JD, Jones SP, Keeley A, Keseru GM, Klein HF, Martin MP, Noble MEM, O'Brien P, Powell A, Reddi RN, Skyner R, Snee M, Waring MJ, Wild C, London N, von Delft F, Walsh MA (2020). Nat Commun 11, 5047-5047.
- 9ZM0 Data: Atg23 Interacts With Both the N- and C-termini of Atg9 Via a Hydrophobic Binding Pocket. Bekkhozhin Z, Leary KA, Ragusa MJ (2025). bioRxiv.
- 7QNP Data: Improved Repeat Protein Stability by Combined Consensus and Computational Protein Design. Michel E, Cucuzza S, Mittl PRE, Zerbe O, Pluckthun A (2023). Biochemistry 62, 318-329.
![Transition from raw detector images to electron density maps and refined atomic models in Coot. Depicted data is from the 7QNP dataset [14]. The 3D molecule rendering has been produced with the Render Molecule job template, which utilizes the MolecularNodes Blender plugin developed by Brady Johnston.](https://cdn.prod.website-files.com/670f957a3be26367c038245d/69c698806079c5f5265daa1a_thumbnail_v3%20(1).png)



